
What is Ehlers-Danlos Syndrome?
Ehlers-Danlos Syndrome (EDS) is a group of connective tissue disorders that are generally inherited and are varied both in their genetic causes and how they affect the body. The most common characteristics are joint hypermobility (joints that move more than normal), skin hyperextensibility (skin that can be stretched more than normal), and tissue fragility (easy bruising, wounds taking longer to heal and scars that heal abnormally).
Ehlers-Danlos Syndromes are currently classified into 13 subtypes (as of 2017) but by far the most common types are Hypermobile (hEDS) and Classical (cEDS). Each subtype has a set of clinical criteria that help guide with diagnosis.
An individual’s experience with EDS is personal in terms of symptoms and severity and may not necessarily be the same as another person’s experience. Diagnostic criteria exist to help distinguish EDS from other connective tissue disorders, and one type of EDS from another. There are many more possible symptoms for each EDS type than are listed in the official criteria. The symptoms listed here are the most common ones.
What are the symptoms of Ehlers-Danlos Syndromes?
The hallmark clinical manifestations of an Ehlers-Danlos Syndrome are most often joint and skin related and may include:
Joints
Joint hypermobility (they move beyond the joint’s normal range); loose/unstable joints which are prone to frequent spraining, dislocations and/or subluxations (coming out of place but not dislocating); joint pain; early onset of osteoarthritis.
Skin
Soft velvety-like skin; stretchy skin (most associated with Classical EDS); fragile skin that tears or bruises easily (bruising may be severe); severe or unusual scarring; slow and poor wound healing.
Miscellaneous/Less Common
Chronic, debilitating musculoskeletal pain (especially associated with Hypermobile EDS); fatigue/tiredness; gastrointestinal problems (Irritable Bowel Syndrome, etc); mitral valve prolapse, and heart arrhythmia, scoliosis; hernias; poor response to local anaesthetic; arterial/intestinal/uterine fragility or rupture (life threatening and most commonly associated with Vascular EDS).
Other conditions such as POTS (Postural Orthostatic Tachycardia Syndrome – heart rate increases when standing up, often manifesting as fainting or dizziness), blood pressure changes (often related to dysautonomia issues), MCAD (Mast Cell Activation Disorder – sometimes severe and unusual allergy symptoms) are commonly associated with EDS, but are not definitive for diagnosis.
The Issue with our Tissue
Each type of Ehlers-Danlos Syndrome is defined as having a distinct problem in the connective tissue. Connective tissue is the ‘glue’ that the body uses to provide strength and elasticity; normal connective tissue holds strong proteins that allow tissue to be stretched but not beyond its limit, and then safely return that tissue to normal. Connective tissue is found throughout the body: in the skin, muscles, tendons and ligaments, blood vessels, organs, gums and eyes.
In a person with EDS, the connective tissue is not structured normally. Some of, or all of the tissue in the EDS-affected body can be stretched beyond normal limits which can lead to damage. One of the major problems with EDS is that ligaments are too lax to hold the joints correctly in place, and they may dislocate or subluxate. The muscles are then forced to work harder to compensate, and may cause muscle spasms and trigger points (myofascial pain).
A body with EDS can behave unreliably, and issues can be widespread and range in severity, from barely noticeable to life-threatening. It can appear as though a person has a large number of seemingly-unrelated symptoms. This is one reason why medical professionals not familiar with Ehlers-Danlos Syndromes have trouble recognising the varied symptoms as all related to ‘one cause’. It may take decades to get properly diagnosed. Misdiagnoses of Arthritis, Fibromyalgia and Chronic Fatigue Syndrome are common because many of the symptoms, especially joint and muscle pain, chronic fatigue, headaches, autonomic dysfunction, and other symptoms overlap with those of EDS.
What are Hypermobility Spectrum Disorders (HSD)?
Hypermobility Spectrum Disorders (HSD) are a group of conditions related to Generalised Joint Hypermobility (GJH). HSD is diagnosed if a patient has GJH with pain and doesn’t fulfil the criteria for Hypermobile EDS.
How common are Ehlers-Danlos Syndromes?
At this time, research statistics of the Ehlers-Danlos Syndromes show the total prevalence in patients as 1 in 2,500 to 1 in 5,000 people. However, recent clinical experience suggests that Ehlers-Danlos Syndrome may actually be even more common than this. EDS has been called the most under-diagnosed disease in the world (Dr Rodney Graham, UK) so we won’t understand the true prevalence until more undiagnosed patients are identified. EDS affects people of all racial and ethnic backgrounds and is much more common in females than males.
The most common types of EDS are Hypermobile EDS (hEDS) and Classical EDS (cEDS). Vascular EDS (vEDS) is rarer, but considered the most potentially dangerous. Many types of EDS are considered extremely rare.
When do EDS symptoms appear?
Types and severity of symptoms vary for each individual. Although some EDS symptoms can appear in infancy or early childhood, it is more common to start noticing symptoms from the teenage years. Joint pain and dislocations/subluxations are often the first symptoms that start requiring attention.
Many with EDS were once very good at sport or activities that required hypermobility (e.g, gymnastics and ballet) and developed frequent injuries and increasing joint and muscle pain as a result. However, perhaps the most common situation is someone in adulthood who has had pain and other symptoms for many years without answers, or has been diagnosed with other conditions that don’t quite make sense.
What are the types of Ehlers-Danlos syndrome and how are they inherited?
There are thirteen defined types of Ehlers-Danlos Syndrome. Each type is a distinct disorder that ‘runs true’ in a family. In other words, an individual with Vascular EDS will not have a child with Classical EDS.
The chart below lists the thirteen types, whether they are Autosomal Dominant (AD – a child of one parent with EDS has a 50% chance of inheriting EDS) or Autosomal Recessive (AR – both parents must hold a copy of this gene in their genetic inheritance). The genetic basis and proteins/connective tissue that is involved are all known, except for Hypermobile EDS.
| Type of EDS (In order of estimated prevalence) | Approximate Prevalence | Associated Gene(s) | Affected Protein(s) | Inheritance Pattern | Distinguishing Features |
|---|---|---|---|---|---|
|
1 in 3,100 – 5000 |
Unknown |
Unknown |
Autosomal Dominant |
|
|
|
1 in 20,000 – 40,000 |
COL5A1 |
Type V collagen |
Autosomal Dominant |
|
|
|
COL5A2 |
Type V collagen |
||||
|
COL1A1 |
Type I collagen |
||||
|
1 in 100,000 – 200,000 |
COL3A1 |
Type III collagen |
Autosomal Dominant |
|
|
|
COL1A2 |
Type I collagen |
||||
|
Less than 1 in 1,000,000 |
COL1A1 |
Type I collagen |
Autosomal Dominant |
|
|
|
COL1A2 |
Type 1 collagen |
||||
|
Less than 1 in 1,000,000 |
ZNF469 |
ZNF469 |
Autosomal Recessive |
|
|
|
PRDM5 |
PRDM5 |
||||
|
Less than 1 in 1,000,000 |
COL1A2 |
Type I collagen |
Autosomal Recessive |
|
|
|
Less than 1 in 1,000,000 |
TNXB |
Tenascin XB |
Autosomal Recessive |
|
|
|
Less than 1 in 1,000,000 |
ADAMTS2 |
ADAMTS-2 |
Autosomal Recessive |
|
|
|
Less than 1 in 1,000,000 |
PLOD1 |
LH1 |
Autosomal Recessive |
|
|
|
FKBP14 |
FKBP22 |
||||
|
Less than 1 in 1,000,000 |
CHST14 |
D4ST1 |
Autosomal Recessive |
|
|
|
DSE |
DSE |
||||
|
Less than 1 in 1,000,000 |
COL12A1 |
Type XII collagen |
Autosomal Dominant or Recessive |
|
|
|
Less than 1 in 1,000,000 |
C1R |
C1r |
Autosomal Dominant |
|
|
|
C1S |
C1s |
||||
|
Less than 1 in 1,000,000 |
B4GALT7 |
β4GalT7 |
Autosomal Recessive |
|
|
|
B3GALT6 |
β3GalT6 |
||||
|
SLC39A13 |
ZIP13 |
[Source www.ehlers-danlos.com/what-is-eds]
How is Ehlers-Danlos Syndrome and Hypermobility Spectrum Disorder diagnosed?
If you think you might have Ehlers-Danlos Syndrome (EDS) or Hypermobility Spectrum Disorder (HSD), ask your GP if such a diagnosis might fit your symptoms. A double appointment may be helpful to provide adequate time for a detailed discussion. It is quite possible that your GP will have not heard of EDS before, or knows little about it.
Useful resources to bring with you to your appointment:
- International Consortium on the Ehlers-Danlos syndromes in 2017 hosted by Ehlers-Danlos Support UK – GP Toolkit https://gptoolkit.ehlers-danlos.org/?fbclid=IwAR2xRfGXa6YY-DN_jCQhJDDVEc-5M7wnn8L9jhbC2diw_1txFzRUXRJ2dvQ
- American Journal of Medical Genetics ‘The 2017 international classification of the Ehlers- Danlos Syndromes’ https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31552
- (as well as other articles from the March 2017 issue – www.ehlers-danlos.com/2017-eds-international-classification)
Hypermobile EDS Diagnostic Checklist
Since Hypermobile EDS is the most common type of EDS, using the diagnostic checklist is a good place to start. Your GP could work through this checklist with you. Diagnosis starts with your doctor hearing your story. The Beighton Score (see the images below), helps to assess how hypermobile your joints are. The doctor will also examine your skin, and look at your medical history (and that of your immediate family), to determine if other conditions or symptoms might be related. www.ehlers-danlos.com/heds-diagnostic-checklist
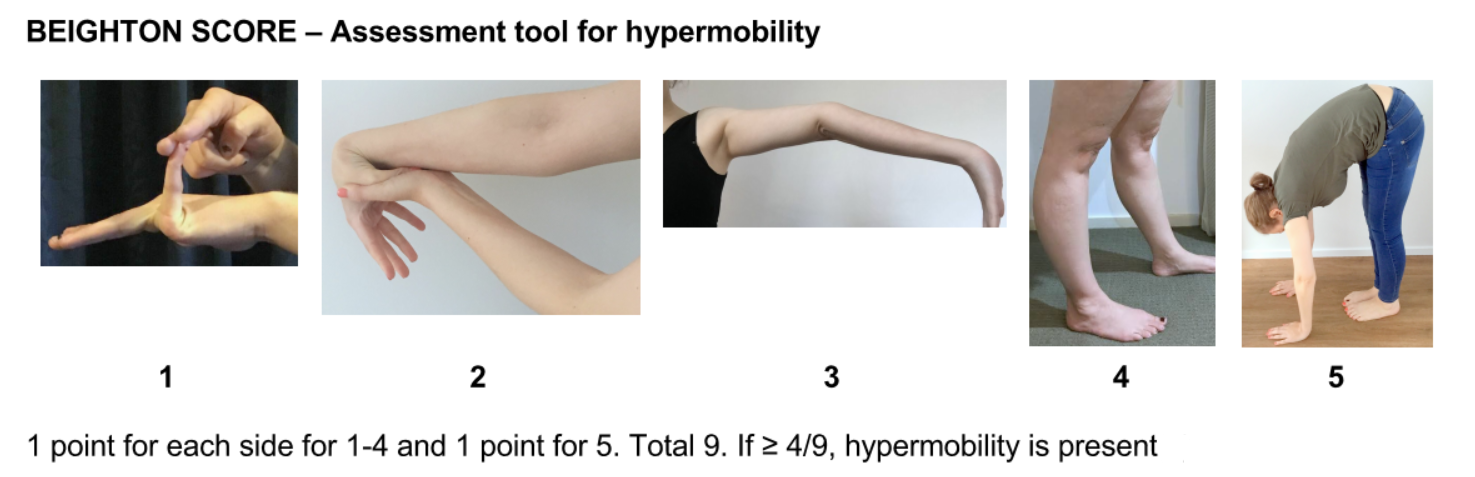
See the useful Beighton Score ‘how to’ video from Ehlers-Danlos Society.
The EDS Clincal Pathway is made for all clinicians in New Zealand who may need some guidance with treatment and diagnosis, this includes GP’s and Physiotherapists. However not all medical professionals are familiar with EDS, and may prefer to refer patients to a specialist to ensure an accurate diagnosis.
In that case, ideally your GP will refer you to a medical specialist with an interest in EDS – usually a rheumatologist, as they may be better able to make a formal diagnosis. There is no guarantee, however, that you will be accepted by the Rheumatology service at your DHB, or that the rheumatologist is experienced with Ehlers-Danlos Syndrome. Awareness and knowledge is growing rapidly, and this is one of the main reasons The Ehlers-Danlos Society NZ exists.
If you do not find adequate help in the public system, there are a few New Zealand rheumatologists in the private sector who specialise in EDS. They will require a written referral from your GP. Some health insurance providers may cover costs.
The Ehlers-Danlos Society NZ is currently building a registry of doctors in New Zealand with training and/or ability to diagnose EDS, and we will post this on the website when available.
The importance of a diagnosis
A correct diagnosis is important because, although EDS/HSD are not curable, many symptoms are treatable. Knowing the type of EDS/HSD gives you and your medical support team some idea of where problems might come from and why they are happening. Prevention of further injury by avoiding certain activities and strengthening of key muscles can also be a helpful strategy once a diagnosis has been made. A diagnosis is also helpful to avoid complications for those patients who may need to consider surgery.
The more patients that are diagnosed with EDS will help the EDS movement worldwide. As the numbers increase, more education and awareness will happen, increasing the likelihood of expanded research that might lead to finding key treatments, and even a cure.
Can I get genetic testing in New Zealand?
A definitive diagnosis for all EDS subtypes (except for hEDS) is ideally made with a genetic test. In New Zealand however, genetic testing in the public system is usually reserved for suspected Vascular EDS patients. Outside of Vascular EDS, the public health system will often not fund genetic testing. Fortunately in EDS types other than Vascular EDS, a formal clinical diagnosis is often adequate for most people to facilitate a practical initial management plan.
What is the prognosis of someone with an Ehlers-Danlos Syndrome?
The prognosis depends on the type of Ehlers-Danlos Syndrome and the individual patient. Life expectancy can be shortened for those with Vascular Ehlers-Danlos Syndrome due to the possibility of organ and vessel rupture. Life expectancy is usually not affected in other types. There can be a wide or narrow range of severity within a family, but each person’s case of Ehlers-Danlos Syndrome will be unique. While there is no cure for the Ehlers-Danlos Syndromes, there is treatment for some symptoms, and there are preventative measures that can be very helpful.
Why the Zebra?
The Zebra is the international EDS mascot. Usually when you hear hoof beats, people assume the common Horse instead of the rarer Zebra. Each Zebra has unique stripes, just as every person with EDS has their own individual set of symptoms. Interestingly, a group of Zebras is called a ‘dazzle’! The Ehlers-Danlos Society NZ has chosen a Kiwi with Zebra stripes as our mascot, so that we are at one with EDS patients internationally, but are also uniquely Kiwi.
Information from Ehlers Danlos Society.